It was very well received. The last time we had this much interest in a talk was in 2005 after we presented the SOLiD sequencer at AGBT and I was a veteran of that crowd. This was my first attendance to the International Cannabinoid Research society and I wasn’t expecting such a welcoming reception.
To understand how you might react to a particular drug or better understand your predisposition to various diseases, sequencing of the coding content of your genome is the best bang for your buck. This is termed Exome sequencing and prices vary from $1000 to $15,000 for such work in 2013. The difference in cost is generally related to 3 things.
1)How much work you want to do scrubbing all of the data at the end (this thread is one example of where you could end up)
2)If the lab is CLIA and CAP certified and
3)If the lab offers a genetic counselor to help interpret the work.
Direct to Consumer tests (DTC) are usually cheaper and offer less interpretation. The FDA has expressed some concern over these tests but we are of the opinion that your genome is yours to read. If what you do with the information inflates the cost of the socialized medical care system… then you probably won’t be free to do it for long unless you pay for it yourself. Late in 2013, the FDA sent a cease and desist letter to 23&Me. We disagree with this over-reaction from the FDA and support 23&Me’s efforts. Their product reflects the freedom for each individual to read their own genome without a government body obstructing that basic human right. They are the Kahn Academy of genomics and their work has shed light on Parkinsons Disease while serving as a patient community message board. Their product alone has informed high networth individuals of potential inherited risks and hence brought significant funding to the field of genomics.
Direct to Physician Tests (DTP) are often more comprehensive and do not attempt to ‘dumb down’ the technical jargon for ease of reading. This is an important point as part of the reason for this blog post is that I have found errors in my 23 and Me exome and there are currently limited tools to contact the company and inform them of this error. There is no option for “contact us” about your test results. The relationship stops with questions. This is not uncommon for low cost DTC tests as the companies are handling 100,000s of samples and there are not enough genetic counselors in the country (3500 estimated) to handle this load.
This is a story about taking a very affordable $999 Exome test from 23 and Me and the diagnostic odyssey it induced. This is likely an extreme example and I have heard of many successful examples of exomes curing patients and believe in this science but feel obligated to highlight shortcomings so they do not pollute the field. In the interest of being open with conflicts, I am involved in a business which sequences patients in a DTP manner called Courtagen Life Sciences. We do not currently offer exome sequencing as we have been waiting for their quality to improve to meet our clinical standards (currently missing an exon in every gene or 1 in 20 bases and they collapse many pseudogenes in mapping). We see substantial improvements in exome quality in 2013, but we do not compete in the DTC markets. We use these ‘drafting’ exomes as a reflex tool to pan for genes which our targeted neurology panels are not designed to cover. We prefer this tiered approach as the targeted panel sequencing is higher quality today particularly with genes which have a lot of pseudogene counterparts or Paralogs. This is the case in many neurological diseases as well as mitochondrial disease. Here is a publication of ours which highlights the importance of deep sequencing across the mitochondria genome. Finally many Exome kits fail to target the mitochondria to depth sufficient to call sub percentages of heteroplasmies.
To highlight this technical detail, we have 1,196 genes in our NucSEEK panel, 415 of which have 1513 pseudogene copies in the genome. Many of these pseudogenes have 500bp stretches of DNA which are identical. Some are not just gene-pseudogene Paralogs but instead negative regulators of the other (ATAD3A and ATAD3B). Exomes capture 100-300bp fragments so there is no way for an Exome to differentiate the source of the DNA between ATAD3A and ATAD3B. Techniques which can capture longer DNA molecules (MIP or LR-PCR) can dance around this problem more effectively by targeting neighboring unique sequence and cleanly segregating pseudogene DNA from clinical DNA but are usually more expensive. Nevertheless, exomes are still a very valuable and are an affordable panning tool given the user understands these short comings.
Interpretation of the 80,000 variants one expects from an Exome can lead to alot of false positives. The best practice is to leverage the phenotypes of the patients to weigh the data in light of the clinical context. This is the golden rule to avoid the incidentilome. Weeding through these incidental findings is where experts are currently needed and in short supply. Incidental findings are the majority of what you will find with such tools and there is grave concern in the genetics field that Exome sequencing is soon to become the next whole body MRI fad known to induce more and more unnecessary testing. The socialized nature of healthcare implies one size fits all federal rules will soon be applied to personalized medicine if we are not careful in our efforts to limit false positives. This obvious contradiction of socialization versus personalization runs the risk of destroying the best tool to come along to healthcare since antibiotics (next gen sequencing) so we must be vigilant in pointing out questionable techniques.
My family has both a maternal and a paternal history of heart disease. My Father was diagnosed with lone paroxysmal atrial fibrillation in his early 40’s. He failed multiple anti-arrhythmic drugs. Stress, alcohol and caffeine appeared to be reliable triggers for the arrhythmia. Ultimately, he was placed on combination of beta blocker and Flecainide (a class I C anti-arrhythmic medication) with reasonably good control of his arrhythmia. (Interestingly there is a 23 and Me variant in their genotyping panel which may have informed this beta blocker hunt (rs1801253 described by Liggett SB et al. (2006)) 30 years ago.)
My brother Brendan, also at age 40 has been diagnosed with atrial arrhythmias. Ambulatory monitoring documented frequent bursts of atrial fibrillation and atrial tachycardia. Trans-telephonic monitor ordered by his physician also documented a short run of wide complex tachycardia which may represent atrial fibrillation with aberrancy vs. Nonsustained Ventricular Tachycardia. Family members currently carry Alive Cor monitors which enable EKG recordings from an iPhone or Smart Phone. They are $200 but you can’t get one unless a Doctor orders it for you! Having built an EKG for an MIT bioengineering class, I have a deep respect for noise and how well this handheld device handles the problem. Here is an EKG recorded from my fingertips while holding the iPhone.
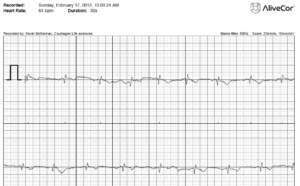
AliveCor ECG taken from fingerprints on an iPhone 4s
Fortunately for us, my sister, Melissa is an Electrophysiologist and was paying close attention to our brothers care. This reminded me that I had my Exome sequenced before and perhaps I should look into it to see if any cardiac gene mutations were present which might play a role in the mechanism for arrhythmias. In addition to this, Partners Health was running a clinical trial on whole genome sequencing and heart disease and Brendan qualified for the study. We contacted the PI (Dr. Patrick Ellinor) and provided him our exome data.
A 3rd benefit we had in our favor was that the three McKernan brothers happen to run a CLIA certified DNA sequencing facility (Courtagen) with Dr. Richard Boles as our medical director. Both Dr. Boles and Dr. Ellinor (Brendans Doctor) saw 2 mutations in the 23 and Me Exome report which caught their attention. Both mutations described genes involved in heart disease. So how severe were these mutations? Are they homozygous? Are they predicted to impact protein function and what does the evolutionary conservation of these nucleotides look like? All very important questions we’ll get into in a moment.
The 23 and Me reports are included here. The Original report was revised in Oct 2012.
This is where things get interesting. The 2 mutations of interest were RYR2 and DSP. An experienced Clinical DNA sequence scientist will notice the reports fail on many levels.
First, neither mutation of concern was in the original report and their reason for showing up in the second report isnt very clear other than to state that other exomes were considered in annotating the final report. This is becoming a popular way to annotate exomes but I believe it is one of the reasons for the false positives in my report.
Several other concerns,
1)The reports should be sorted on gene name so compound heterzygotes can be easily highlighted.
2)Compound hets should be clearly itemized and attempts to phase them with bioinformatics or sequencing of the parents should be recommended.
3)Heterozygosity or homozygosity is not spelled out in the report. This is critical.
4)Not all genes are created equal. Some exhibit dominant phenotypes others recessive and this should be spelled out when describing risk of the mutation.
5)As a customer who has had their whole family tested at 23 and Me, there is no report which compares your exome to previous genotypes performed at 23 and Me to confirm or refute sample mix ups or errors. Families should be checked for mendelian errors.
6)How damaging are the mutations? The reports do make a call on this but its not clear the algorithms are reviewed in a clinical context. Any one algorithm can have many blind spots.
So before we get too excited over these potential ‘smoking guns’, we should fill in the short comings in the 23 and Me report. Luckily we have these tools as standard analysis tools for all variants tested at Courtagen and I was able to put these two variants through those tools. We used 5 different algorithms to weigh in on the severity of the nonsynonymous mutations.
SIFT, Mutation Taster, Mutation Surveyor, PolyPhen2, Grantham Scores
All of these prediction tools labeled the RYR2 mutation as Highly damaging. DSP being a frameshift mutation will also be highly damaging but again we are uncertain if either of these mutations are homozygous or heterozygous and if they dominant or recessive genes.

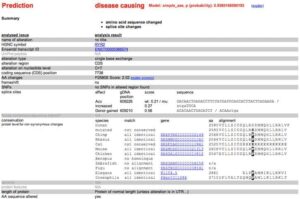
Even with the use of 5 mutation prediction tools we do not have a 100% sensitivity while many of them fail to consider the evolutionary conservation of the amino acid. Doing this we see this amino acid is conserved all the way to Lamprey implying Darwin does not like it when this base changes.

Highly conserved Amino Acid back to Lamprey
Naturally if this is so damaging, we need to understand if I am a homozygote, or a heterozygote for this mutation and if my Brother shares the same mutation.
Being vesting in this outcome, I personally stepped in and sequenced these 2 mutations in our CLIA certified clinical lab. We designed 2 PCR assays for each variant and perform forward and reverse sanger sequencing on on each assay across myself and Brendan (16 sanger reads in total) and none of the Sanger data confirmed the reported variants. These were labeled as high quality variants with 23 and Me and they are clearly wrong according to the Sanger data. I initially believed the Sanger data as the reads are longer and less prone to mismapping while multiple primer designs confirm the event with bidirectional Sanger sequencing. Requesting multiple primer designs does not always safeguard against all artifacts (foreshadowing intended).
An excerpt of that data is here but to summarize, NONE of the 16 sanger reads validate 23 and Me’s findings.
SANGER DATA CAN BE DOWNLOADED HERE-
A program to view Sanger trace files can be downloaded from 4Peaks. The sequence information from these files can be aligned to the human genome using a program called BLAT from Jim Kent at UCSC. I aligned these reads to both Hg18 and Hg19 to ensure the discrepancies were not due to database version numbers and coordinate cross talk.
The Sanger Amplicons were also run on an Agilent HS Biolanalyzer to ensure there was one primary peak for each amplicons and that pseudogenes or off target PCR was not playing a role in the Sanger Sequencing. Below is an in-silico overlay of 4 amplicon peaks.
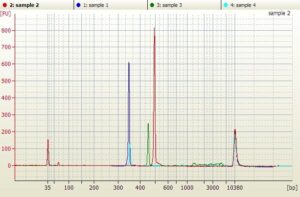
In our experience it is rare for ILMN data to not match Sanger data. What can cause this discrepancy?
This could be a sample swap but this would be identifiable by comparing the data to previous 23 and Me genotypes. To our knowledge, this has not been done yet but many of the mutations exist in both the April and the October reports. It would be helpful to have the genotype concordance with previous tests!
This could also be related to mismapping and structural aspects of the sequence. The DSP variant, does exist but only as a silent mutation, not a frame-shifting insertion as seen below.
Its possible the secondary structure of this sequence was challenging to sequence with bridge PCR. Below is a mfold analysis of the secondary structure and one can see a very long inverted repeat which could present substantial sequencing challenges.
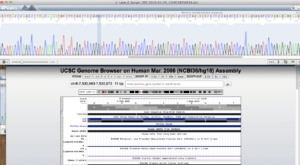
Below is the BLAT alignment of my RYR2 Sanger Read to Hg19. Base 237,814,721 should be a variant.

Variants from DTC exomes should be Sanger Validated or validated with second platform in a certified CLIA lab before deeming them ‘Actionable’. This is likely to cost $100-$200 and could save you a wealth of time, concern and unnecessary doctor visits. The selection of which variants to Sanger Validate is best reviewed in light of the clinical context of the patient.
Two reports were generated by 23&Me and only the second report had these putative false positives. The second report claimed to use population based genotyping where one persons genotypes are compared to all exomes on hand to improve the results. Although this is a good idea in theory I think its dangerous if not deployed properly as common variants and systematic sequence errors will be shadowed onto other peoples genomes. The reasons for my concerns are rooted in my experience developing next generation sequencers and my intimate understanding of how easily SNP calls can change by slight alterations in read length or chemistry versions. If one doesnt properly separate samples which were sequenced with different versions of the chemistry (75bp reads vs 101bp or 10 cycles of PCR vs 8 cycles of PCR) dramatic differences in FPs can be observed. If one is using population based genotyping, many of these systematic errors will be propagated from one version of the chemistry onto another. I suspect this is what at play in this case of FPs as other people at 23&Me have reported these same variants and the dbSNP frequency is very low for these.
The next steps in this odyssey for me is to sequence Brendans Exome myself. This is being done on 2x250bp reads on a MiSeq to use the longest reads possible for the price to avoid mis-mapping. In addition I used Nextera library construction techniques which are completely enzymatic manipulation of the DNA. In my experience, Focused acoustic bombardment, nebulization, and end repair are significant sources of error in sequencing and fully enzymatic methods are less damaging.
My only concerns are the reported lower Q20 coverage stats over targets these capture reagents report but this maybe addressable with a 20Gb exome.

Cycle 267 on a MiSeq Nextera Exome
Here is an excel sheet 23 and Me FPs with potential False Positives in the 23 and Me data. We do not see the RYR2 false positives with the paired 250bp from the MiSeqs using Haloplex sequencing of the RYR2 gene in other patients. This gene is in our NucSEEK panel. Results Pending on our RYR2 mutations.
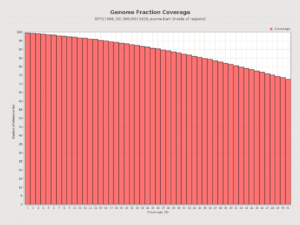
We completed a HaloPlex 37Mb Exome. At 1400 mm^2 density we hit 83X coverage with 1 Miseq run. The Halo Exome was designed for 100bp reads and the average insert size is 150bp for paired 100mers. The MiSeqs 250bp reads are arguably wasteful for this type of sequence but still provide the best results for small population of fragments which are larger than this.
Nevertheless, the coverage of a single MiSeq Run with 2x250bp reads is quite good.
VCF file recapitulates the RYR2 mutation but also exposes 2 in phase variants upstream. These variants may have impacted the Sanger Assays amplification if primers overlapped the these two novel variants. This implies 23 & Me’s Exome data for one of the variants in question was more reliable than the Sanger data.

Variants upstream of RYR2 mutation of interest are in phase with the novel variant and could induce allele specific amplification or FN Sanger confirmations
Further exome sequencing has confirmed the RYR2 mutation in Brendan, Kevin, Brian and our Mother. The DSP variant remains an error. This does highlight the risks of SANGER primer design in validating any novel mutation. Upstream private mutations which are not in dbSNP should be considered in the primer design. Most importantly, this has implications for exome capture techniques which do not tile a target region. Haloplex has at least 8 amplicon tiles across a given target. Techniques like Raindance which uses primer design similar to a Sanger pipeline may fall victim to private mutation allele drop out.
The best study on mutational burden in RYR2 receptors is from Argelia Medeiros-Domingo. Of note in this paper is how common polymorphisms when compounded can induce calcium channel leaks. In our case we have both a heterozygous private mutation (P2566S in exone 51) and a 14% frequency mutation (Q2958R in exon 61). The common polymorphism is homozygous in our father and heterozygous in the children while our father does not have the private P2566S mutation. My father has Arrythmias so the RYR2 private mutation cannot be the sole arrhythmia causing allele in our family as this mutation is maternal.
Quoting,
We have learned that common polymorphisms in other ion channels have the potential to
modify the clinical phenotype22 ,23 ; polymorphisms in RYR2 may have the same potential.
RyR2-Q2958R is the most common RYR2 polymorphism; was described for the first time 9
years ago24 and is particularly common in Caucasians (34%). The second most common
polymorphism in RYR2 is G1886S (20% African Americans, 9% Caucasians) followed by
G1885E (6% Caucasians). Interestingly, in vitro studies in heterologous systems have
demonstrated that both G1885E and G1886S polymorphisms caused a significant increase in
the cellular Ca(2+) oscillation activity compared with RyR2 wild-type channels. Further, when
both polymorphisms were introduced in the same RyR2 subunit, the store-overload-induced
calcium release activity was nearly completely abolished25 . The clinical consequences of this
“RyR2 loss of function” in vitro phenotype is not clear, however, compound heterozygosity
involving these two polymorphisms has been reported in right ventricular dysplasia26 . The
potential functional effects of the 6 novel polymorphisms identified in this study are unknown.
It is important to remark that none of the novel mutations detected on this study have been
functionally characterized to further bolster the contention of pathogenicity. However, less
than 15% of the mutations reported to date in RYR2 have been studied in vitro, pathogenicity
has been suspected based on co-segregation with the disease and absence in control subjects.
Here, co-segregation with the disease data was not available for all cases. Instead, the
prevalence of putative mutations amongst strong cases (~60%) was markedly higher than in
controls (~3%) and all putative mutations were absent in 400 reference alleles. Thus, although
the precise contribution of each discrete mutation to the phenotype remains to be determined,
statistically, the estimated probability for pathogenicity for RYR2 mutations found in strong
cases is quite high (~95%).
Comprehensive open reading frame mutational analysis of RYR2
Chr1,
| P2566S | Missense/cSNP |
Chr1,
| Q2958R | Missense/cSNP |
The ESP6500 project has recently released over 178 missense mutations for RYR2. The P2566S mutation does not exist in this dataset either.
Variants in RYR2 which cause calcium leaks have been reported to cause Atrial Fibrillation in Mouse models.
RYR2 calcium leak drives AFib in Mouse
Knowing we may have found candidates for our maternal arrhythmias in the family, we began investigating mutations in our exomes which might explain our paternal contribution.
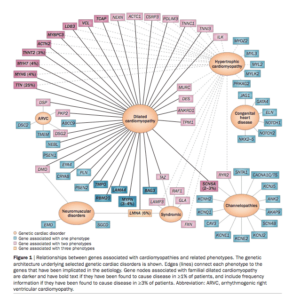
We narrowed this search to 58 genes known to be involved in Cardiac disease and plan to expand this gene list search in time as more literature on arrhythmias matures.
| ACTC1 |
| ACTN2 |
| ANK2 |
| CACNA1C |
| CACNB2 |
| CASQ2 |
| CAV3 |
| CRYAB |
| CSRP3 |
| DES |
| DMD |
| DSC2 |
| DSG2 |
| DSP |
| EMD |
| FBN1 |
| GLA |
| GPD1L |
| HCN4 |
| JPH2 |
| JUP |
| KCNE1 |
| KCNE2 |
| KCNH2 |
| KCNJ2 |
| KCNQ1 |
| LAMP2 |
| LDB3 |
| LMNA |
| MYBPC3 |
| MYH6 |
| MYH7 |
| MYL2 |
| MYL3 |
| MYOZ2 |
| PDLIM3 |
| PKP2 |
| PLN |
| PRKAG2 |
| RYR2 |
| SCN4B |
| SCN5A |
| SGCA |
| SGCB |
| SGCD |
| TAZ |
| TCAP |
| TGFB3 |
| TGFBR2 |
| TNNC1 |
| TNNI3 |
| TNNT2 |
| TPM1 |
| TTN |
| VCL |
| STIM1 |
| TMEM43 |
| TTN |
Further exome sequencing has also highlighted a CASQ2 mutation in Brendan (rs4074536) which has been published on by Wang et al. CASQ2_RYR2_Mutations. Kevin and Brian do not have this mutation but do share a 5’UTR VUS just upstream of this N-Terminal mutation. Further exome sequencing is being performed in the rest of the family to see if the CASQ2 mutation tracks with the Paternal line phenotype but the Wang paper implies this exact mutation is 42% population frequency and likely benign.
| 196 A>G | T66A | Missense/cSNP |
CACNA1C presents 3 homozygous Amino Acid changing SNPs.
Brendan and Brian are Homozygous for all 3. Kevin is homozygous for K1893R and heterozygous for the other two.
All three of these are predicted to be benign by Provean. One of them being a Methionine had me stumble into the following paper (CACNA1C_Exonic_Promoters) which describes how there are alternative Transcriptional Start sites on the C-terminus of CACNA1C and that these make 3 different transcripts. The TSS appear to be similar coordinates as our Methionine mutation. This paper was published in April of 2013 underscoring the importance of yearly re-annotation of exomes.
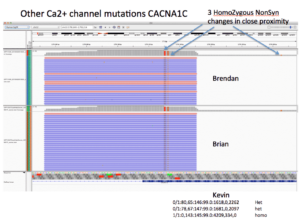
CACNA1C Homozygous mutations are located near an alternative TSS and impact a Methionine
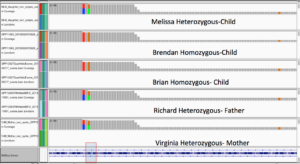
Family Exomes Show heterozygous mutation profile for most SNPs in the parents and homozygous variants for Brendan and Brian suggesting a different penetrance for 2 siblings.
The alternative TSS sites are showcased in this figure from the above paper on Exonic Promoters.
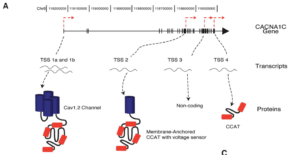
Exons 40-47 participate as Alternative exon promotors for other gene products. “Benign” Methionine changes need to be reconsidered in light of this.
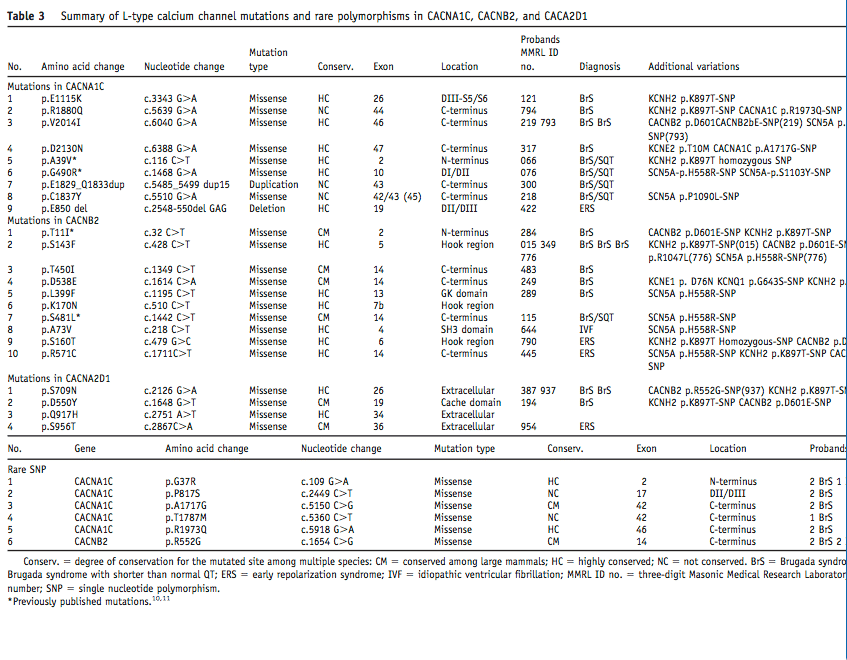
Its clear from the literature that CACNA1C mutations can present variable phenotypes. The Diagnosis column in Table 3 in the Burashnikov paper highlights how mutations indifferent exons of the gene can create completely different phenotypes. There is not only internal promoters as shown above but there is also alot of literature showcasing alternative splicing creating diversity in calcium signalling, sensitivity to certain calcium blockers and interactions with RYR2 and Calmodulin.
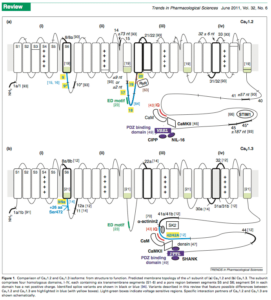
Exons 41-43 are important for CaM binding. In fact this paper highlights Diltiazem sensitivity with certain splice isoforms which can induce Bradycardia (an episode experienced by Brendan). Also notice below the RYR2 interaction domain below.

Although there is ample smoke with these mutations and the plethora of literature associating mutations in these genes causing rhythm problems, its clear channelopathies can produce a spectrum of unexpected outcomes particularly when protein:protein interactions are at play across multiple genes in the same pathway. A molecular model would be ideal but I suspect difficult to build in a mouse model with 3 highly differentially spliced 40+ exon genes with over 6 variants. The myriad of alternative splicing events which are tissue specific may complicate moving this model to a mouse but would certainly be interesting. Ramon Brugada suggested we make IPS cells and coax them into cardiomyocytes to see if we can replicate this effect in vitro. Anyone with experience with this and interested in the topic feel free to email me at Kevin dot McKernan at courtagen dot com.
What is clear from the literature is that compound “thought to be independently benign” mutations when combined in the same gene or in different genes in the same pathway are reported to cause disease.Compound mutations-a common cause of severe long qt syndrome-Circulation-2004-Westenskow-1834-41
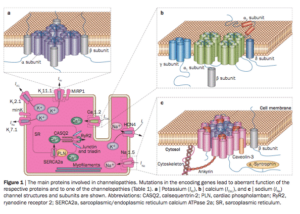
RYR2, CASQ2 and CACNA1C mutations are known to create Calcium leaks which can induce Arrhythmias. Calcium and Beta Blockers have resulted in Bradycardia for most family members. Other calcium pathway targets need to be investigated. ARMGO pharmaceuticals is investigating Rycals. These are in phase II trials and likely unavailable. Their website has a nice video on the impact of calcium leaks and the preferred mechanisms of Rycals. http://www.youtube.com/watch?v=F6Kmz3-cZMo
Other mechanisms of choice which are more available- Ryan et al demonstrates how Mitochondrial Calcium is regulated by Cannabidiol (CBD) which is a non psychoactive cannabinoid synthesized by Cannabis. Despite it being classified as a cannabinoid, CBD has very weak affinity for CB1 and CB2 receptors. See Non-Psychoactive Cannabinoids. Literature today points to CBD being a moderate PPARgamma agonist. Cannabidiol_Esposito_PPARgamma.
Key to understanding this pathway is a background on the Mitochondrial Permeability Transition Pore or mPTP. This is a protein pore in the inner membrane of the mitochondria which when open allows 1500 Dalton molecules or smaller enter the inner membrane of the mitochondria. Often this is triggered by higher intracellular calcium levels and this pores prolonged opening will lead to apoptosis.
Quoting wikipedia
Various factors enhance the likelihood of MPTP opening. In some mitochondria, such as those in the central nervous system, high levels of Ca2+ within mitochondria can cause the MPT pore to open.[20][21] This is possibly because Ca2+ binds to and activates Ca2+ binding sites on the matrix side of the MPTP.[6] MPT induction is also due to the dissipation of the difference in voltage across the inner mitochondrial membrane (known as transmembrane potential, or δψ). In neurons and astrocytes, the contribution of membrane potential to MPT induction is complex, see.[22] The presence of free radicals, another result of excessive intracellular calcium concentrations, can also cause the MPT pore to open.[23]
Other factors that increase the likelihood that the MPTP will be induced include the presence of certain fatty acids,[24] and inorganic phosphate.[25] However, these factors cannot open the pore without Ca2+, though at high enough concentrations, Ca2+ alone can induce MPT.[26]
Stress in the endoplasmic reticulum can be a factor in triggering MPT.[27]
Effects of MPT[edit source | editbeta]
Multiple studies have found the MPT to be a key factor in the damage to neurons caused by excitotoxicity.[7][8][6]
The induction of MPT, which increases mitochondrial membrane permeability, causes mitochondria to become further depolarized, meaning that Δψ is abolished. When Δψ is lost, protons and some molecules are able to flow across the outer mitochondrial membrane uninhibited.[7][8] Loss of Δψ interferes with the production of adenosine triphosphate(ATP), the cell’s main source of energy, because mitochondria must have an electrochemical gradient to provide the driving force for ATP production.
In cell damage resulting from conditions such as neurodegenerative diseases and head injury, opening of the mitochondrial permeability transition pore can greatly reduce ATP production, and can cause ATP synthase to begin hydrolysing, rather than producing, ATP.[31] This produces an energy deficit in the cell, just when it most needs ATP to fuel activity of ion pumps such as the Na+/Ca2+ exchanger, which must be activated more than under normal conditions in order to rid the cell of excess calcium.
MPT also allows Ca2+ to leave the mitochondrion, which can place further stress on nearby mitochondria, and which can activate harmful calcium-dependent proteases such ascalpain.
Reactive oxygen species (ROS) are also produced as a result of opening the MPT pore. MPT can allow antioxidant molecules such as glutathione to exit mitochondria, reducing the organelles’ ability to neutralize ROS. In addition, the electron transport chain (ETC) may produce more free radicals due to loss of components of the ETC, such ascytochrome c, through the MPTP.[32] Loss of ETC components can lead to escape of electrons from the chain, which can then reduce molecules and form free radicals.
MPT causes mitochondria to become permeable to molecules smaller than 1.5 kDa, which, once inside, draw water in by increasing the organelle’s osmolar load.[33] This event may lead mitochondria to swell and may cause the outer membrane to rupture, releasing cytochrome c.[33] Cytochrome c can in turn cause the cell to go through apoptosis(“commit suicide”) by activating pro-apoptotic factors. Other researchers contend that it is not mitochondrial membrane rupture that leads to cytochrome c release, but rather another mechanism, such as translocation of the molecule through channels in the outer membrane, which does not involve the MPTP.[34]
Much research has found that the fate of the cell after an insult depends on the extent of MPT. If MPT occurs to only a slight extent, the cell may recover, whereas if it occurs more it may undergo apoptosis. If it occurs to an even larger degree the cell is likely to undergo necrotic cell death.[9]
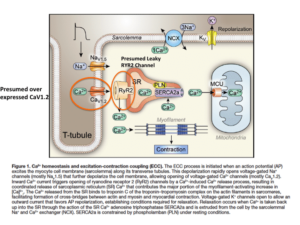
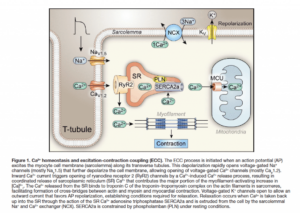
CACNA1C is a cellular calcium channel, where RYR2 is an organelle calcium channel found mostly in the Sarcoplasmic or endoplasmic reticulum. Mutations can cause calcium leaks in these receptors which are known to cause cardiac arrhythmias. Andrew Marks is well published on the RYR2 receptor.
Other suggested reading-
Modified
UnModified
What is the connection between PPARgamma and calcium? It is known that PGC-1alpha (PPAR Gamma CoActivator alpha) stimulates mitochondrial biogenesis. The attached paper highlights the connection to calcium regulation.
3.1 Calcium- and second messenger-dependent
regulation of PGC-1a expression
Increased contractile activity translates into a sustained
increase in intracellular calcium concentration, which activates
the calcium-dependent phosphatase calcineurin
(CaN) and Ca2fl-calmodulin dependent kinase (CaMK).35
CaN controls the expression of PGC-1a in muscle, providing
a calcium-signalling pathway whose stimulation results in
increased mitochondrial content.36 Loss of calcineurin in
heart results in impaired mitochondrial electron transport
associated with high levels of superoxide production.37
Moreover, in early embryonic development, the dephosphorylation
of nuclear factors of activated T cells (NFATs) by calcineurin
is required for mitochondrial energy metabolism.
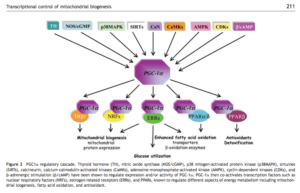
PPARgamma pathway
Interestingly, Ryanodine receptors have also been reported to be involved in the pathway for synthesis of endocannabinoids and this also involves calcium.
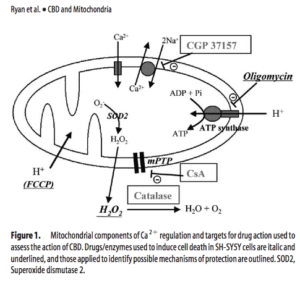
To add further support to Ryan’s conclusions, O’Sullivans manuscript titled “Time-dependent vascular actions of cannabidiol in the rat aorta” demonstrates CBD directly impacts calcium levels and likely does this via PPARgamma and PPARgamma independent mechanisms. Recent work linking the Mito VDAC1 gene to direct CBD influence would support this work.
Ethanol is a reproducible trigger for the family members after age 40. Dondas et al showcase how EtOH can bind to L-Type and Ryanodine receptors and impact calcium flow. Likewise Fatjo demonstrate how alcoholics have increased expression of L-type calcium channels. Presence of cardiomyopathy decreases this expression.Ethanol_caffeine_impact_on_RYR2_L-type_channels

CBD is known to inhibit FAAH and increase serum anandamide levels (AEA). These Molecular targets for CBD are also described in Bisogno et al.
Recently many papers have highlighted AEA anti-arrhythmia properties. Kury et al demonstrate AEA effect on voltage-dependent sodium and calcium channels in rat ventricular myocytes.
Knowing CBD is a candidate to regulate calcium imbalance in the mitochondria what is its safety profile and how can it be procured?
Studies in 1980 measured 400mg chronic does of CBD for Epilepsy patients and found the compound to very safe and well tolerated. Bergamaschi also revisited the safety question in 2011 coming to similar conclusions.
Table 2 has human studies up to 600mg with no effects on heart rate or blood pressure .

What is the legal status of CBD? Many compounds from the Cannabis plant are schedule I labeled yet Hemp milk and <0.3%THC products are legally sold at Whole Foods.
From Wikipedia
Legal status[edit source | editbeta]
Cannabidiol is not scheduled by the Convention on Psychotropic Substances.
The legal status of cannabidiol in United States is unclear. In Schedule I[58][59] there is a broad category called “Tetrahydrocannabinols”, although cannabidiol is not chemically a tetrahydrocannabinol. Cannabidiol has DEA statistical number 7372,[60] but it does not necessarily mean it is illegal.
Cannabidiol is a Schedule 2 Drug in Canada.[61]
20 States now have medical cannabis laws and most of these dispensaries carry CBD edible products. Smoking is not recommended due to the impact on the cardiovascular system. Vaporization, edibles, capsules or tinctures are preferred.
Dixie Botanicals also sells CBD capsules (25mg). Eventhough CBD has a wide therapeutic index many papers point to biphasic dose response curves suggesting knowing your dose is important. Several companies produce analytical review of Dixie’s products. Its important to see that their CBD is half activated. The plant makes CBDA or carboxylated CBD and this is decarboxylated with heat to CBD.
http://analytical360.com/m/edibles/69634
There have been few papers studying the difference between CBDA and CBD. Here is one which details the plants synthetic pathway for making both molecules.
After 2 months of CBD treatment, Brendan’s arrhythmias have ceased. He visited his physicians at MGH, told them the story and they took great interest in the pathways and mechanisms we have been studying.
Dosage started at 12mg/day or two 1ml droppers (6mg/ml). He moved this up to 3 times a day as he was having arrhythmias at night and in the morning before dosage. Its expected to have a 6 hr clearance and three 12mg doses a day has stabilized his symptoms. CBD is currently expensive and we have been looking at other sources. Unfortunately some have peppermint oil which has some history of inducing arrythmias so we approaching those with caution at this time and will post relevant literature as we dig into this. This underscores the importance of QC on products in this field. We have heard of various anecdotal cases of products from Dixie being tested at second labs and not matching their advertised CBD ratio. Since Epilepsy patients have recently shown great success with CBD treatment, there has been a run on CBD and it is backordered frequently.
Other family members are looking into this as well.
Limitations of this data: 100s to 1,000s of different genetic defects can likely all share the common phenotype of arrhythmias. This data does not support the use of CBD for any arrhythmia. This successful treatment is likley specific to this private case of calcium channel mutations and caution is advised extending this treatment to any person with the relatively vague descriptor of having “arrhythmias”.
We would like to thank Lester Grinspoon and Raphael Mechoulam for helpful discussions and providing the related manuscripts above. We would like to thank Ramon Brugada for review of the work as well.
Disclaimer. All of this sequencing was performed using D.R.E.AM-PCR. This method will debut in Nature Biotechnology on Tuesday Oct 8th 2013. DREAM PCR stands for Decontamination Ready Encoded Amplification. The method generates ephemeral PCR products and is contamination proof. It also uses 5 bases in PCR thus the encoding ensures our amplicons do not match anything in the USPTO. This is important as 20% of human genes are believed to be patented (Jensen Murray).
ASHG, Boston 2013
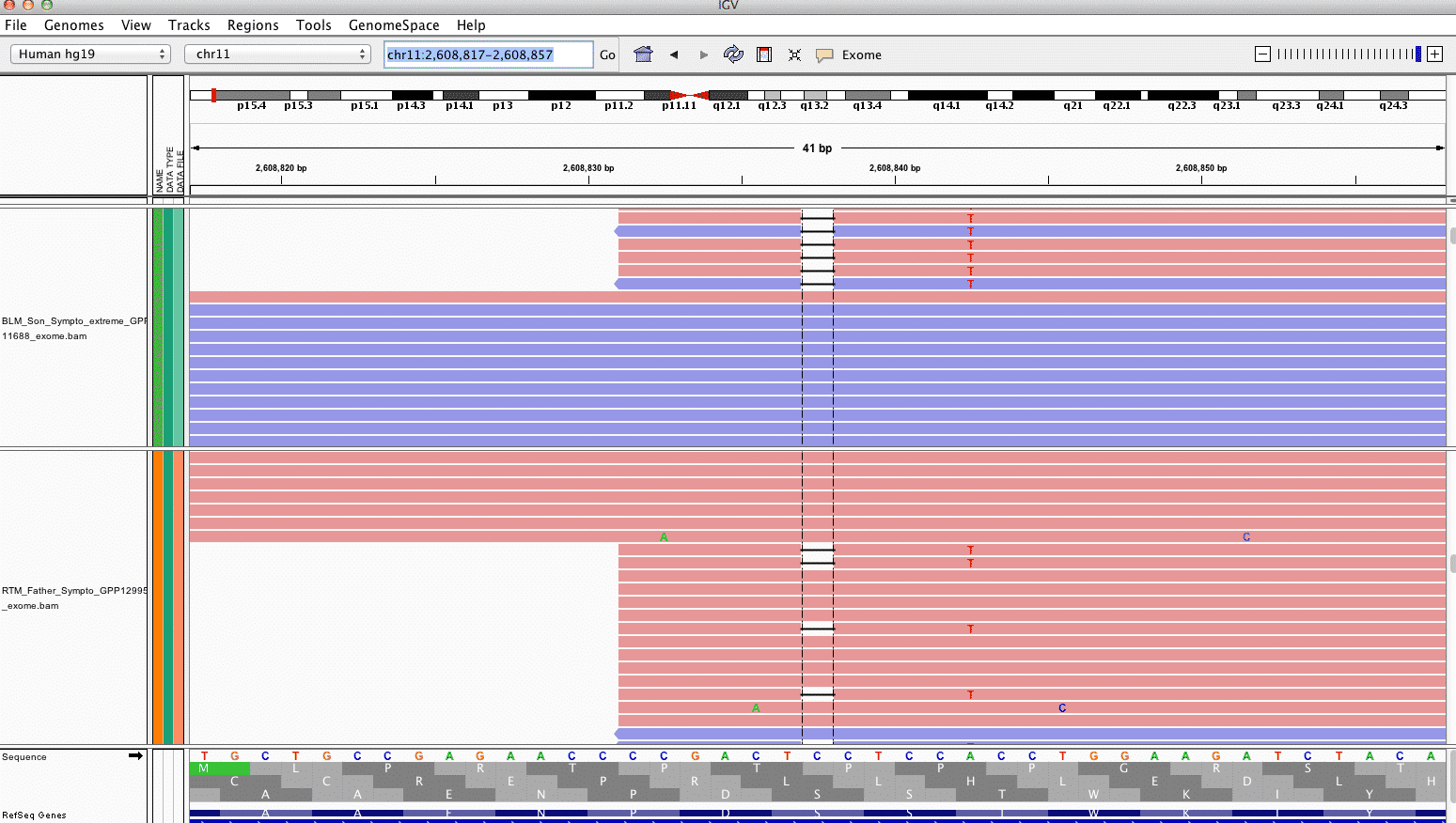
KCNQ1 has graduated as an interesting variant after having Ingenuity review this data. After further review of this frameshift variant in IGV it appears to be an adaptor trimming artifact (only seen in amplicons with one start point) and has been dismissed.
With the recent excitement over CBD in Epilepsy, there has been a shortage of available CBD from most suppliers. This has led to Brendan going off of CBD only to see his Arrhythmias return. Although this is a great control it is not the desired way to live considering the publications in this space regarding cardiac sudden death. We have also become aware of these shortages inducing poor quality control in the products which has let us to TLC QC. Ideally this would be performed with HPLC but TLC is lowest cost system to spot check product. Software for quantifying images can be uploaded here.
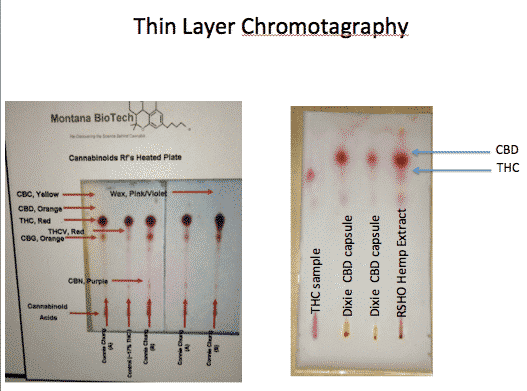
Profile from the above software

Patch Clamp treatment of demonstrate Anandamide reducing arrhythmia
{kind=link}
{kind=link}